Dr Wendell Zhang
Disclaimer
This is an educational piece and is not intended to provide advice in this complicated field. Please refer to your local guidelines and treating team for specific details regarding perioperative management strategies for patients.
Key points
-
Disorders of coagulation are uncommon but can have a significant impact on perioperative optimisation for patients
-
All patients with coagulopathies are at higher risk of developing intraoperative or postoperative haemorrhage
-
There are specific subgroups of patients who can develop severe coagulopathies as a result of their pathologies
-
There are multiple different options for risk mitigation in the emergency setting for patients
-
The use of fresh frozen plasma is not supported as first-line therapy for patients with coagulopathy
Preamble

You are an anaesthetic trainee assigned to the emergency list. In front of you is Mr Green, a 60-year-old man awaiting a laparotomy for small bowel obstruction. Glancing through his notes, you see Mr Green has Haemophilia A and has had previous surgeries needing a comprehensive haematological plan. You ring the hospital haematologist and start putting together a plan to avoid catastrophic haemorrhage.
A brief overview of haemostasis
Haemostasis comprises complex, intersecting and concurrent pathways. The end goal of haemostasis is to keep blood where it is supposed to be – inside vessels. To that end, the key outcomes are as follows:
-
Primary Haemostasis: Platelet accumulation at the site of injury to form a “platelet plug”
-
Secondary Haemostasis: Reinforcing the platelet plug with a “fibrin plug”
Central to both processes is the activation of large amounts of thrombin. While these processes tend to happen concurrently, it is helpful to break them down into various steps.
There are two key biochemical coagulation pathways (the extrinsic and intrinsic pathways) which converge upon the common pathway to produce thrombin and ultimately, fibrin.
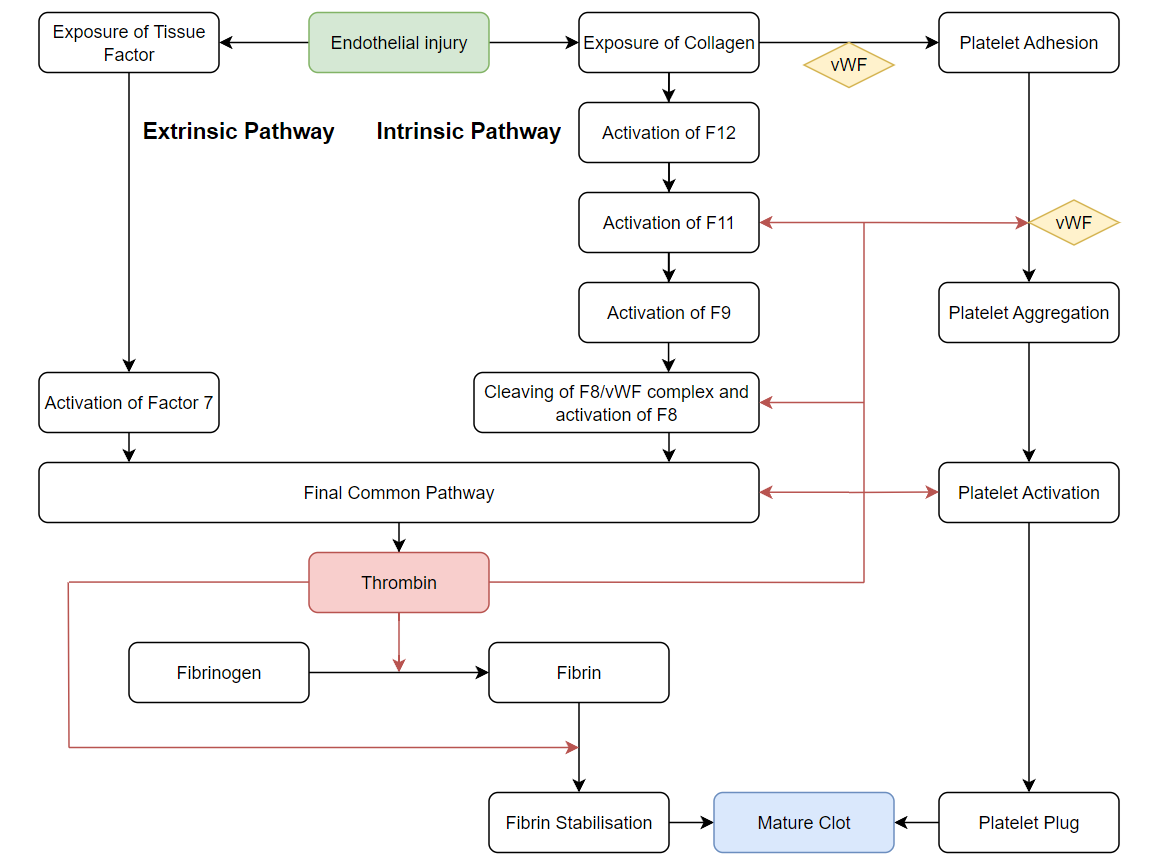
Tissue Injury and Primary Haemostasis
The initiating step for haemostasis is the exposure of blood to sites of endothelial damage or foreign material. Collagen and tissue factor exposed at the site of damage attracts Von Willebrand’s Factor (vWF) which creates a surface conducive to platelet adhesion.
Simultaneously, the exposure of tissue factor initiates the extrinsic pathway. Factor 7 is activated by tissue factor which in turn activates the final common pathway (which directly generates thrombin).
Once platelets adhere to vWD-collagen complex, platelet activation commences, resulting in structural changes that increase surface area for platelet adhesion. They also release prothombotic agents, chiefly adenosine diphosphate (ADP), thromboxane and thrombin, which augment further platelet activation and drive positive feedback loops to culminate in mass platelet aggregation. Finally, activated platelets upregulate surface receptors, including GIIb/IIIa, which form crosslinks via locally secreted vWB and fibrinogen with other platelets to ultimately form the “platelet plug”.
Secondary Haemostasis
Secondary haemostasis is the process which culminates in fibrinogen being converted to fibrin by the actions of thrombin, thereby creating a fibrin plug to stabilise the clot.
Fibrinogen is found in large quantities in the blood so the limiting step is typically the availability of thrombin. Thrombin is initially rapidly generated by the extrinsic pathway as mentioned previously. In addition to aiding in the activation and adhesion of platelets, thrombin is key to propagating positive feedback loops for the intrinsic pathway.
The intrinsic pathway is slower and activated by contact from the blood with exposed collagen or other negatively charged particles. One of the key steps mediated by thrombin is the cleaving of vWF from F8 and subsequent activation of F8.
Furthermore, the body needs large quantities of thrombin to facilitate the creation of the fibrin plug. Thrombin generation acts as a positive feedback mechanism for itself as it increases the availability of F5 which is a cofactor in the activation of prothrombin to thrombin . In addition, thrombin also activates further F13 which facilitates the cross-polymerisation of fibrin into a stable clot.
|
Key Clotting Factors |
Pathway |
Synthesis |
|
1 (Fibrinogen) |
Common |
Liver |
|
2 (Prothrombin) |
Common |
Liver – Vitamin K dependent |
|
3 (Tissue Factor) |
Extrinsic |
Damaged cells |
|
4 (Calcium ions) |
All |
Plasma |
|
5 |
Common |
Liver |
|
7 |
Extrinsic |
Liver – Vitamin K dependent |
|
8 |
Intrinsic |
Platelets |
|
9 |
Intrinsic |
Liver – Vitamin K dependent |
|
10 (Thrombokinase) |
Common |
Liver – Vitamin K dependent |
|
11 |
Intrinsic |
Liver |
|
12 |
Intrinsic |
Liver |
|
13 (Fibrin Stabilising Factor) |
NA |
Liver, platelets |
Inherited Coagulopathy
|
Disorder |
Factor Affected |
Inheritance |
Prevalence |
|
Haemophilia A |
8 |
X-linked recessive |
1 in 5,000 males |
|
Haemophilia B |
9 |
X-linked recessive |
1 in 30,000 males |
|
Haemophilia C |
11 |
Autosomal Dominant/Recessive |
1 in 100,000 |
|
Rare Factor Deficiencies |
F1,2, 5, 7, 10, 13 |
Autosomal Recessive |
Less than 1 in 1,000,000 |
|
Von Willebrand’s Disease |
vWF |
Autosomal Dominant/Recessive |
1 in 10,000 |
|
Glanzmann’s Thrombasthenia |
Platelets |
Autosomal Recessive |
Less than 1 in 1,000,000 |
|
Bernard-Soulier Syndrome |
Platelets |
Autosomal Recessive |
Less than 1 in 1,000,000 |
Haemophilia and Factor Deficiencies
Haemophilia is a group of diseases characterised by deficiencies of various clotting factors.
Haemophilia A is an X-linked recessive disorder (primarily affecting males) that is caused by a deficiency in F8. It is the most common variant of haemophilia and is typically detected in early childhood.
Haemophilia B is likewise an X-linked recessive disorder affecting F9. It is less common and typically less severe than haemophilia A.
Haemophilia A and B can arise as a de novo mutation with a significant portion of patients having no family history of the disease. Women may rarely present with these diseases by inheriting mutations from both parents. Some heterozygous carriers may also present with milder variants of the disease due to varying inactivation of the X-chromosome and lyonisation. Both diseases can cause haemarthrosis and intramuscular haematomas.
Haemophilia C is a very uncommon autosomally inherited disorder of F11. Unlike haemophilia A and B, it is inherited with an autosomal pattern. A key differentiating factor is that patients with haemophilia C do not develop haemarthrosis.
Patients with haemophilia may present with a prolonged APTT (though a normal APTT does not exclude haemophilia). Definitive diagnosis is typically achieved by specific clotting factor assay demonstrating significant deficiencies. The severity of haemophilia A and B is graded by the percentage activity of the affected factors on a clotting factor assay. The severity of haemophilia C and the patient’s risk of bleeding are not necessarily associated with its relative factor deficiency.
|
Severity of Haemophilia |
Factor Activity |
|
Mild |
5-40% activity |
|
Moderate |
1-5% activity |
|
Severe Disease |
<1% activity |
Other specific factor deficiencies (F1,2,5,7,10,13) do exist but are exceedingly uncommon.
Von Willebrand’s Disease and Platelet Disorders
Von Willebrand’s Disease is caused by a qualitative or quantitative deficiency of Von Willebrand’s factor.
There are three main variants of this disease:
-
Type 1 has reduced quantity of vWF
-
Type 2 has qualitative defects in vWF
-
Type 3 has a severe quantitative deficit of vWF
It is characterised by prolonged mucosal bleeding and easy bruising. Patients may have a prolonged APTT due to increased degradation of F8 but a normal APTT does not exclude this disease. Further quantitative (von Willebrand’s Factor antigen) and qualitative studies (von Willebrand’s factor activity) can further differentiate patients into the disease subtypes.
Platelet-specific disorders such as Glanzmann’s Disease and Bernard-Soulier Disease are very uncommon diseases which affect platelet aggregation and adhesion respectively.
Preoperative Assessment

Time permitting, it is important to establish the type and severity of a patient’s coagulopathy before any surgical procedure.
History
A focused history from the patient may include a history of previous heavy menstrual bleeding, mucosal bleeding, intra-articular/intramuscular haematomas or easy bruising. It is also important to establish prior procedural history (surgeries, dental procedures), and whether any of these were complicated by bleeding and if the patient has previously received directed therapy or blood product transfusions for bleeding. It may also be relevant to establish a family history of bleeding disorders.
Recency of bleeding events, their severity, and need for intervention also provide an indication of stability of disease and whether specialist haematology input is required to optimise bleeding risk.
Examination
In addition to a standard preoperative assessment, it may also be important to establish features suggestive of liver disease, and vitamin C/K deficiency. A thorough examination may also reveal evidence of current mucosal, intramuscular, intraarticular, or subcutaneous bleeding
Investigations
On top of basic pathology investigations before an operation, further blood tests may shed light onto the specific mechanisms of a patient’s coagulopathy. Additional testing may include coagulation studies with mixing studies and specific factor levels. In addition, patients should have a blood group and hold in anticipation for the potential need for blood products intraoperatively. If patients have had previous blood transfusion there is a risk they may have minor antibodies. These can potentially cause significant delays in finding compatible blood.
Risk Mitigation

Multidisciplinary Discussion
For complex patients with coagulopathies, it is often important to have a multidisciplinary discussion with other specialities to plan for a safe operation. These patients will warrant a discussion with a hospital haematology service for further expertise and specific advice regarding preoperative management.
With the surgical and theatre staff, it would be important to discuss the exact timing of the operation. For instance, it may be important to clarify the urgency of the operation and if delays can be facilitated for further investigations and patient optimisation. In addition, operations which may be complicated would ideally be performed during normal daytime hours.
For smaller surgical centres, there may be limitations on the supply of medications and specific blood products. These may need to be ordered in advance or alternative therapies may be indicated in the acute setting. In addition, it is important to establish with the hospital pathology service what investigations would be feasible to perform at short notice (such as factor assays), noting the delay for samples to be transported to tertiary centres.
Preoperative optimisation for Haemophilia
|
Therapeutic options |
Pros |
Cons |
|
Recombinant Factor |
|
|
|
High Purity Factor |
|
|
|
Cryoprecipitate |
|
|
|
Prothrombin Complex Concentrate |
|
|
|
Desmopressin |
|
|
|
Tranexamic Acid |
|
|
|
vWF and F8 concentrate |
|
|
Preoperative optimisation for patients with haemophilia is geared around mitigating the primary causes of morbidity and mortality in this group. These are intraoperative blood loss, intracranial haemorrhage and the transmission of blood-borne infections.
To limit intraoperative and postoperative bleeding, patients are typically provided with aggressive correction of their deficient clotting factor, typically aiming for between 80-100% correction. In the postoperative period, the target levels are not as high but patients will typically still require ongoing factor replacement for several weeks. Evidence from the United Kingdom demonstrates significant challenges in achieving target levels for haemophilia B patients in the perioperative and postoperative setting.
There are various therapeutic options for replacing deficient factors.
Recombinant Factor is essentially comprised of pure clotting factor and is the preferred mode of replacing clotting factor. It does not put the patient at increased risk of contracting a blood-borne infection and there are well-defined dosing guidelines to achieve clotting factor targets. However, it is quite expensive and not every hospital service will have access to this resource.
High and intermediate purity factor concentrates are blood products refined from plasma and treated to minimise the risk of blood-borne infections. It is more readily available and cheaper than pure recombinant factor but does carry a minor risk of blood-borne infection transmission.
Cryoprecipitate, as the name suggests, is frozen plasma which is thawed and placed in a centrifuge with the precipitate being isolated as the end product. It contains fibrinogen, F8, F13 and vWF. It is commonly available in hospitals as it is also frequently used with massive transfusion protocols. However, cryoprecipitate administration places patients at risk of transfusion reactions and should not be used unless no other therapies are available.
Fresh Frozen Plasma is comprised of whole plasma, containing all clotting factors and no platelets. It is no longer routinely used for perioperative optimisation of patients with coagulopathy given its risk of adverse transfusion reactions, fluid overload and the existence of superior therapeutic options.
Preoperative Optimisation for Von Willebrand’s Disease
Similar to haemophilia, patients with Von Willebrand’s Disease will require perioperative optimisation with the aim of correcting their coagulation deficits either by promoting the release of VWF or by providing external vWF and F8 replacement.
Desmopressin (DDAVP) is a synthetic variant of the endogenously produced vasopressin. In addition to its anti-diuretic actions, it also acts to stimulate the release of vWF from endothelial cells. Importantly, DDAVP is only used in Type 1 VWD (low levels of vWF production). It is not used in Type 3 VWD (no factor produced) and is used with caution in Type 2 VWD (only certain sub-variants have a desirable response to this therapy).
Tranexamic acid promotes clot stabilisation and is typically used for patients with VWD and haemophilia. There is a hypothetical risk of increased thromboembolic disease but this has not been demonstrated in clinical trials. It should be avoided in patients with genitourinary bleeding as this can cause urinary retention secondary to clot formation.
Patients with VWD (Especially those with Type 2 and 3 VWD) may benefit from VWF and F8 concentrates in combination. There is a risk for adverse reactions in individuals who develop alloantibodies to vWF
Anaesthesia technique:
In this population group with inherited coagulopathy, it is generally advisable to minimise tissue trauma. Particular care should be taken with essential procedures such as intravenous, and intraarterial access, as well as the insertion of airway devices.
The benefits and risks of local anaesthesia should be evaluated carefully in this patient group. If local injections are used, the use of a vasoconstrictor together with the local anaesthetic may minimise bleeding. Spinal, epidural and paravertebral blocks are relatively high-risk procedures for this patient group and a comprehensive risk benefit discussion will be needed for neuraxial techniques.
Drugs which may increase the risk of bleeding such as NSAIDs should be avoided in these patients. Patients should have mechanical DVT prophylaxis with careful consideration regarding the role of pharmacological DVT prophylaxis postoperatively.
Conclusion
Inherited Coagulopathies present a rare but significant challenge to anaesthetists in the perioperative setting. Great care should be taken in this patient group and perioperative planning will be a complex multidisciplinary endeavour.
References
Shah UJ, Narayanan M, Graham Smith J. Anaesthetic considerations in patients with inherited disorders of coagulation. Continuing Education in Anaesthesia, Critical Care & Pain. 2015 Feb 1;15(1):26-31.
Martlew VJ. Peri-operative management of patients with coagulation disorders. British journal of anaesthesia. 2000 Sep 1;85(3):446-55.
O’Donnell JS, Lavin M. Perioperative management of patients with von Willebrand disease. Hematology 2014, the American Society of Hematology Education Program Book. 2019 Dec 6;2019(1):604-9.
Hazendonk HC, Preijers T, Liesner R, Chowdary P, Hart D, Keeling D, Driessens MH, Laros‐van Gorkom BA, van der Meer FJ, Meijer K, Fijnvandraat K. Perioperative replacement therapy in haemophilia B: an appeal to “B” more precise. Haemophilia. 2018 Jul;24(4):611-8.
Membership of the Working Party:, Harrop‐Griffiths W, Cook T, Gill H, Hill D, Ingram M, Makris M, Malhotra S, Nicholls B, Popat M, Swales H. Regional anaesthesia and patients with abnormalities of coagulation: the association of anaesthetists of great britain & ireland the obstetric anaesthetists’ association regional anaesthesia UK. Anaesthesia. 2013 Sep;68(9):966-72.